
SMARTPOP Manual
2 Requirements
3 Installation
4 SMARTPOP features
4.1 Input
4.1.1 Via command lines
4.1.2 Via input files
4.1.3 Windows executable
4.2 Simulation parameters
4.2.1 Verbose
4.2.2 Random seed
4.2.3 Population size
4.2.4 Sample size
4.2.5 Number of simulations
4.2.6 Number of generations to run
4.2.7 Mating system and number of offspring
4.2.8 Sibling matings
4.2.9 Demography parameters
4.2.10 Mutation rates
4.2.11 Burn-In phase
4.2.12 DNA sequences
4.2.13 Choice of outputs
4.2.14 Save/Load
4.3 Outputs
4.3.1 Diversity tables
4.3.2 Fasta
4.3.3 Arlequin
4.4 Running SMARTPOP in parallel mode
4.5 Setting up a complex scheme (change of parameters through time)
4.6 Starting conditions: burn-in and pre-run
5 Examples
A Default parameters
B References
1 Introduction to SMARTPOP
SMARTPOP is fast and flexible forward-in-time simulator for population genetics. Specially developed for speed efficiency, it is available in both serial and parallel versions. Developed for anthropological inferences on human populations, SMARTPOP simulates individuals with sequences of sex-linked DNA (mitochondrial, X and Y chromosomes) and autosomes. Studies of social dynamics are enabled using SMARTPOP's flexible demographic models and social rules of mating.
For any use of SMARTPOP or re-use of its code source please cite:
Guillot and Cox, 2014. SMARTPOP: inferring the impact of social dynamics on genetic diversity through high speed simulations. BMC Bioinformatics 2014 15:175
2 Requirements
SMARTPOP has been developed in C++ using the Boost C++ library. To build the software from sources you need a C++ compiler such as g++ or Visual Studio installed on your computer. To compile the parallel version of SMARTPOP, we recommend using mpic++.
3 Installation
You can directly download a binary (executable) version of SMARTPOP compatible with your OS at http://smartpop.sourceforge.net/download.html
Alternatively you can build SMARTPOP from the source code following these instructions:
To build SMARTPOP from source on a UNIX machine:
- Download the source code from http://smartpop.sourceforge.net/download
wget http://smartpop.sourceforge.net/download/SMARTPOP.zip - Move the archive to the folder where you want SMARTPOP to be installed
mv SMARTPOP.zip /home/login/foo - Uncompress the archives
unzip SMARTPOP.zip - Move to directory
cd SMARTPOP/src-serial/ - Build SMARTPOP
make - Optional: Build the test package
make test - Optional: Run the test package
./test
SMARTPOP is then ready to launch via the command line:
./smartpop
For building the parallel version, go to the src-parallel directory:
- cd SMARTPOP/src-parallel/
- make
- Launch the parallel version (on UNIX system) on four cores:
- mpiexec -n 4 ./smartpopMPI
We have run tests on the main operating system available today, if you encounter any trouble please contact us.
4 SMARTPOP features
SMARTPOP must be called via the command lines (./smartpop ) from the directory where it is installed (or add the directory to your PATH). One call will launch a set of simulations with one set of parameters.
4.1 Input
A set of simulations rely on several parameters, all of which have default values except the population size and random seed. The default values are given in this table. You can either set these parameters using flags on the command line or via an input file.
4.1.1 Via the command line
You can define all the parameters to start a set of simulations using flags on the command line. All the parameters that are not called by flags will receive default values.
Note: you must always define the population size.
The order of parameters does not matter. If you give a wrong sequence of arguments on the command line, the program will raise an error and not start (in most cases).
To run a set of 1000 simulations with population size 200 evolving for 500 generations, enter:
./smartpop -p 200 -t 500 -nsimu 1000
| Flag | Argument |
Meaning |
| -v |
Verbose | |
| -i |
Input file with parameters | |
| -o | string |
Name for the output files |
| -s | integer |
Random seed |
| -p | integer |
Population size |
| -sample | integer |
Sample size |
| -nsimu | integer |
Number of simulations |
| -t | integer |
Number of generations between two sampling events |
| -nstep | integer |
Number of sampling events through time for one simulation run |
| -mat | integer |
Mating system |
| -noSib |
Prevent half siblings (sharing at least one parent) mating | |
| -noSib2 |
Prevent full siblings (sharing two parents) mating | |
| -mu | double (x4) |
Mutation rates (simple rates) |
| -muFull | double (x8) |
Mutation rates (Kimura’s two parameters model) |
| -mtdiv |
Output diversity for mitochondrial DNA | |
| -ydiv |
Output diversity for Y chromosome | |
| -xdiv |
Output diversity for X chromosome | |
| -adiv |
Output diversity for autosomes | |
| -nbLociA | integer |
Number of unlinked autosomal loci |
| -sizeA | integer |
Size, in number of sites, per autosomal locus |
| -nbLociX | integer |
Number of unlinked loci on the X chromosome |
| -sizeX | integer |
Size, in number of sites, per locus on the X chromosome |
| -sizeY | integer |
Size, in number of sites, on the Y chromosome |
| -sizeMt | integer |
Size, in number of sites, of the mitochondrial sequence |
| -burnin | double |
Set the diversity threshold parameter for the burn-in phase |
| -burninType | int |
Set the type of DNA to be tested in the burn-in phase |
| -demog | double (x3) double |
Set the demographic function |
| -fasta |
Save each simulation in a fasta format file at the end of the run | |
| -arl |
Save each simulation in a arlequin format file at the end of the run | |
| -save | string |
Save all the simulations in a SMARTPOP format (.sim) with a root name given by the argument |
| -load | string |
Load all the simulations in a SMARTPOP format (.sim) from the root name given by argument |
| -header |
Output the header at the beginning of diversity files | |
4.1.2 Via input files
Instead of defining the parameters in the command line, you can also use an input file. For example, this is useful if you modify a lot of default value parameters.
You must respect the format of this file to make it work. Each line describe the value for one parameter. The order of lines does not matter. All the parameters that are not defined in this input file will take default values, given in this table.
| Keyword | Argument |
Meaning |
| verbose | boolean (0 or 1) |
Verbose |
| fileOutput | string |
Name of the output files |
| seed | integer |
Random seed |
| populationSize | integer |
Population size |
| sampleSize | integer |
Sample size |
| nSimu | integer |
Number of simulations |
| step | integer |
Number of generations between two sampling events |
| nstep | integer |
Number of sampling events through time for one simulation run |
| matingSystem | integer (0 to 4) |
Mating system |
| inbreeding | boolean (0 or 1) |
0 allows siblings mating;1 prevents half sibling matings; 2 prevents full sibling matings |
| muMtDnaTransition | double |
Mutation rate (/site/generation) for mitochondrial transitions |
| muMtDnaTransversion | double |
Mutation rate (/site/generation) for mitochondrial transversions |
| muXTransition | double |
Mutation rate (/site/generation) for X chromosome transitions |
| muXTransversion | double |
Mutation rate (/site/generation) for X chromosome transversions |
| muYTransition | double |
Mutation rate (/site/generation) for Y chromosome transitions |
| muYTransversion | double |
Mutation rate (/site/generation) for Y chromosome transversions |
| muAutosomeTransition | double |
Mutation rate (/site/generation) for autosomal transitions |
| muAutosomeTransition | double |
Mutation rate (/site/generation) for autosomal transversions |
| diversityToOutput | integer (0 to 4) |
0 = output diversity for all kinds of DNA simulated |
|
1 = output mitochondrial DNA | ||
|
2 = output X chromosome | ||
|
3 = output Y chromosome | ||
|
4 = output autosome | ||
| nbLociA | integer |
Number of unlinked autosomal loci |
| sizeA | integer |
Size, in number of sites, per autosomal locus |
| nbLociX | integer |
Number of unlinked loci on the X chromosome |
| sizeX | integer |
Size, in number of sites, per locus on the X chromosome |
| sizeY | integer |
Size, in number of sites, of the Y chromosome |
| sizeMt | integer |
Size, in number of sites, of the mitochondrial sequence |
| burninTheta | double |
Set the diversity threshold parameter for the burn-in phase |
| burninType | int |
Set the type of DNA to be tested in the burn-in phase |
| demog | double double double |
Set the demographic function |
| mito | integer |
Mitochondrial simulation only |
| fastaOutput | boolean (0 or 1) |
Save each simulation in a fasta format file at the end of the run |
| arlequinOutput | boolean (0 or 1) |
Save each simulation in a Arlequin format file at the end of the run |
| save | boolean (0 or 1) (+ string) |
If 1, save all the simulations in a SMARTPOP format (.sim) in the directory given by the argument |
|
If 0, no saving | ||
| load | boolean (0 or 1) (+string) |
If 1, load all the simulations in a SMARTPOP format (.sim) from the directory given by the argument |
|
If 0 no saving | ||
| headerOutput | boolean (0 or 1) |
Output the header at the beginning of diversity files |
By default, each run of SMARTPOP creates a parameter file called SMARTPOP_parameters.txt. This has exactly the same format as the input parameter file such that running ./smartpop -i SMARTPOP_parameters.txt will run the exact same set of simulations as you formerly ran. This is possible due to the fact that the random seed is one of the input parameters.
4.1.1.3 Windows executable
The available windows executable only handle input files. SMARTPOP will read the file
4.2 Simulation parameters
4.2.1 Verbose
It is recommended to begin using SMARTPOP with the verbose option on. This will make SMARTPOP return relevant information one the command line when the simulations are running. It is a good way to check that the parameters are set correctly, as well as to visualize the progress of the program.
4.2.2 Random seed
Simulations are highly reliant on random processes. Such processes are simulated via sequences of random numbers which are a large part of the program. Each time the program starts, it calls a random seed from which this sequence is produced uniquely. By default, the random seed is random, but it is possible to set it to repeat earlier runs.
4.2.3 Population size
The population size set in SMARTPOP corresponds to the census population size at the beginning of the simulation. If the population size is set to be non constant, this will change through time.
4.2.4 Sample size
Instead of analyzing the entire population, you can sample a certain number of individuals. This situation would match a “real life” situation where you do not have access to the DNA of your whole population. If you define a sample size, all the outputs (diversity, but also fasta and arlequin files) will be generated on a random sample in your population of the defined size.
4.2.5 Number of simulations
You can define the number of simulations that will be run with this set of parameters. If you are loading a set of simulations from a directory, this number must be smaller or equal to the number of saved simulations.
4.2.6 Number of generations to run
During each simulation, the population will evolve for a number of generations t between two sampling events. By default, there is only one sampling event at the end of those t generations, where output files are produced and diversity is measured. If you define multiple sampling events (NSampling) through time, then t defines the number of generations to run between two sampling:
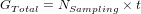
4.2.7 Mating system and number of offspring
Five mating systems are available, each designated by a number:
- Monogamy
Males and females are paired randomly to mate. No individuals can be paired with two different mates. The number of offspring per couple is set to follow a Poisson law. - Polygamy
Males and females are paired randomly to mate. The number of offspring per female is set to follow a Poisson law. - Polygyny
Males and females are paired randomly to mate. A female can only mate with one male. The number of offspring per female is set to follow a Poisson law. - Polyandry
Males and females are paired randomly to mate. A male can only mate with one female. The number of offspring per male is set to follow a Poisson law. - Random mating
Males and females are paired randomly to mate. There is no law on the number of offspring.
4.2.8 Sibling matings
For any mating system, you can forbid the mating between full or half siblings.
4.2.9 Demography parameters
The demograpy is defined by a Markovian function:
popsize(t+1)=a + b x popsize(t) + c x popsize(t)2
Based on three parameters a, b and c, this definition permits a large range of demographic scenarios. This table presents the most simple example:
| Scenario | a | b | c |
| A constant population size | 0 | 1 | 0 |
| A constant growth of x individuals per generation | x | 1 | 0 |
| A constant decrease of x individuals per generation | -x | 1 | 0 |
| An exponential growth of rate x | 0 | x | 0 |
| An exponential decrease of rate x | 0 | 1/x | 0 |
4.2.10 Mutation rates
Each type of DNA (mitochondrial, X, Y and autosome) has two mutation rates: a transition and transversion rate. These rates must be set in mutations/site/generation. Overall mutation rates can be defined, but you can set transitions equal to transversions equal to half the total mutation rate, if you do not know the ratio of transition over transversion.
The rationale behind having only eight mutation rates is that mutation rates measured for mtDNA, Y, X and autosomes can be different by more than an order of magnitude. Similarly, transition rates are usually an order of magnitude higher than transversion rates.
4.2.1 Burn-in phase
By default, simulations will start with the entire population having the same DNA. Alternatively, a burn-in phase allows you to start with an accelerated evolutionary process that will force your population to reach a given diversity θburn-in from which your simulation will start. In this process, a higher mutation rate is applied to your population which quickly increases the diversity. The accelerated evolution stops when the threshold is reached or if it has been running for more than 100 generations.
By default, θburn-in is computed on the mitochondrial DNA. This can be changed by using the flag burnType. This parameter must be set to:
- 1 for mitochondrial DNA
- 2 for X chromosome
- 3 for Y chromosome
- 4 for autosomes
- A mitochondrial sequence
- A sequence from the non-recombining Y chromosome
- A set of unlinked loci on the X chromosome
- A set of unlinked loci on the autosomes
- A file with the simulated DNA (at the end of the run) in fasta format
- A file with the simulated DNA (at the end of the run) in arlequin format [2]
- A file containing the whole population, included individual's DNA in a SMARTPOP format (.sim), that can be read by SMARTPOP.
- A diversity file for each kind of DNA
- A file containing the whole population, included individual's DNA in a SMARTPOP format (.sim), that can be read by SMARTPOP.
- Population size (not the sample size)
- Number of sites in the sequence evaluated
- Number of polymorphic sites
- Proportion of polymorphic sites
- Mean pairwise difference
- Number of haplotypes
- Allele heterozygosity
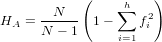
- Nei’s heterozygosity (i.e. heterozygosity averaged per site) [4]

- Theta Watterson θw

- Theta homozygosity θH

- Theta Pi θπ
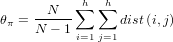
- Tajima’s D
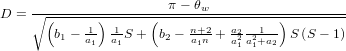
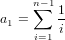
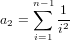
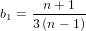
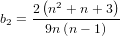
- Assumed equilibrium
It is possible that the population is in a state of equilibrium. The definition of "equilibrium" in population genetics is quite vague, but it is agreed that such equilibrium will be reached by running simulations for a very long time. How long is long enough? It must be at least the TMRCA, to be independent of starting conditions.
As an indicator, the mean and variance of the time to the most recent common ancestor (TMRCA) assuming a constant population size [3, 1, 6] is: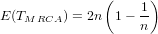
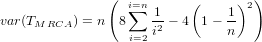
- Burn-In
Alternatively, we offer the user the possibility to start from a known diversity, defined by its θθ. For example one may want to study what happens to a population that had a θθ of 0.2 one thousand generations ago.
This starting point is produced by applying an accelerated mutation scheme on the population. It will assign a mutation rate one order of magnitude higher than the real rate, run some generations and stop once the diversity threshold is reached. The real simulation can then start from this point.
This approach is novel to SMARTPOP. - Make your own recipe
With the flexibility of SMARTPOP, you can choose your own starting scheme. Controlling the mutation rate, you may reach higher diversity than equilibrium. You can also use the demography feature to create your own burn-in phase.
4.2.12 DNA sequences
Each individual has a set of sex-linked DNA sequences:
It is possible to choose the length of each loci by type. Note: all autosomal loci must share the same length; all X loci must share the same length. It is also possible to choose the number of loci on X and autosomes.
For computational reasons, the length of sequences must be a multiple of 32, or it will be automatically forced to the next higher multiple of 32. For instance, if you enter a length of 33, your simulated locus will have 64 sites. The size of the DNA considered unnecessary for a study can be set to 0, making the simulations faster.
4.2.13 Outputs
SMARTPOP can produce different outputs for each simulation:
SMARTPOP also produces output for the whole set of simulations:
If the output name is set to 'foo' in the parameters, then the different files produced would be foo.fasta, foo.arl, foo_div_mt, foo_div_X, foo_div_Y and foo_div_A.
4.2.14 Save/Load
An entire set of simulations can be saved in a directory, and reloaded from the same directory. This allows the construction of complex scenarios with parameters changing through time. It can also be used if you were running very large and long simulations and your server only allows you limited time for each run. In such a case, you would run a succession of “short” simulations, each being stopped and restarted to fit with the scheduler.
Reloading a simulation set you can also chose to sample the previous simulation. For example you can simluate first 1000 simulations and save it. You can then reload those simulation sampling 100 individuals out of it by simply indicating -p 100.
Using the save feature, each population will be written in a SMARTPOP special format (.sim). It is not necessary to understand this format to use the save/load option, but a small description is provided for more complex use of SMARTPOP.
A new file SMARTPOP_XX.sim with all the information needed to reload this population in to the software is written in the current directory when save is activated.
The header of this file is on two lines:
Population_size number_of_female generation size_Mito number_Loci_X size_Per_X size_Y number_Loci_A size_Per_A
mating_system μTransition mtDNA μTransversion mtDNA μTransition A μTransversion A μTransition X μTransversion X μTransition Y μTransversion Y
There after each line represents an individual. The first number indicates the sex (1 for female, 0 for male) following by numbers representing its DNA.
The DNA sequence is written directly as a sequence of 64 bit numbers, so that the file is faster to save and load.
4.3 Outputs
4.3.1 Diversity tables
A file is created for each different type of DNA (mitochondrial, X, Y and autosome). If a name is provided, and the file already exists, the results will append to the end of the existing file.
This file contains different summary statistics for the population for each sample. Each line represent a sample. For each sample, for each DNA type, SMARTPOP will write in the file:
4.3.2 Fasta
You can output the population, or a sample of it, in a fasta format file. Such a file can easily be piped through other population genetics software, such as COMPUTE from the libSequence package[5].
4.3.3 Arlequin
You can output the population, or a sample of it, in a file of the format of ARLEQUIN software. This allows you to make inferences on your simulations using ARLEQUIN [2].
4.4 Running SMARTPOP in parallel mode
It is possible to run SMARTPOP in parallel. You must build a specific version of SMARTPOP using C++ compiler including Message Parsing Interface, such as mpic++.
When running in parallel, the simulations are distributed across several cores at the beginning at the simulation. Each process will write on different files to avoid conflict.
If you save and restart simulations for a complex scenario, be sure to use the same number of cores each time.
4.5 Setting up a complex scheme (change of parameters through time)
A set of simulations launched in one command line has a unique value for each parameter. It is possible to consider a complex scenario, where those parameters change through time, by successive use of SMARTPOP with different parameters.
After each parameter set, the simulations must all be saved. They are reloaded under the new set of parameters. This lets us to link as many successive sets of parameters as desired. The saving / loading process is handled automatically under the flags -save and -load. The same process can be used to load a set of simulation and make it run longer without having to start again from the beginning.
4.6 Starting conditions: burn-in and pre-run
The starting conditions are a fundamental problem in forward-in-time simulations. How to determine the complete genetic set of the population from which the evolution starts? This is equivalent to asking what is the DNA state of an ancient population, which is totally unknown in most studies. In studies based on modern DNA, you have (at most) knowledge about past individuals who left descendant but nothing about the other past members of the population.
The question remains what to start the simulation with. A fully random DNA set is in no way meaningful. Instead the genetic patterns of real population is the result of a long evolutionary process that creates haplotypes - individuals still sharing a large part of their DNA sequence.
The start must therefore be a sequence shared among individuals. From this null diversity, one must produce an artificial diversity, to correspond to a real population. There are three options:
A Default parameters
| Flag + Argument | Input File Keyword + Argument |
Meaning | Parameter File Example | Default Value |
| -v | verbose int |
Verbose | Verbose 1 | 0 |
| -i myfile.txt | NA |
Input file with parameters | SMARTPOP_parameters.txt | NULL |
| -o filename | fileOutput filename |
Name of the outputs | fileOutput smartpop_13145105_2 | smartpop_seed_matingSystem |
| -s integer | seed integer |
Random seed | seed 13145105 | random |
| -p integer | populationSize integer |
Census population size | populationSize 150 | 1000 |
| -sample integer | sample integer |
Sample size for estimation of diversity, and fasta/arlequin outputs | sample 100 | 100 |
| -nsimu integer | nSimu integer |
Number of simulations | nSimu 500 | 500 |
| -t integer | step integer |
Number of generations between two sampling events | step 100 | 100 |
| -nstep integer | nstep integer |
Number of sampling events through time for one simulation | nstep 10 | 1 |
| -mat integer | matingSystem integer |
1 monogamy, 2 polygamy, 3 polygyny, 4 polyandry, 5 random | matingSystem 4 | 2 |
| -noSib | inbreeding 1 |
Prevent half sibling matings | inbreeding 1 | 0 |
| -noSib2 | inbreeding 2 |
Prevent full sibling matings | inbreeding 2 | 0 |
| -mu double (x4) |
Mutation rates (simple rates, respectively mtDNA, X, Y and autosome) | -mu 0.0001 0.0001 0.0001 0.0001 | ||
| -muFull double (x8) |
Mutation rates (Kimura’s model respectively transition mtDNA, transversion mtDNA, transition X, transversion X, transition Y, transversion Y, transition autosome and transversion autosome) | -mu 0.0001 0.00001 0.0001 0.00001 0.0001 0.00001 0.0001 0.00001 | ||
| muMtDnaTransition integer |
Transition rate on mtDna | muMtDnaTransition 0.000001 | 0.000001 | |
| muMtDnaTransversion integer |
Transversion rate on mtDna | muMtDnaTransversion 0.0000001 | 0 | |
| muXTransition integer |
Transition rate on X | muXTransition 0.0000001 | 1e-08 | |
| muXTransversion integer |
Transversion rate on X | muXTransversion 0.0000001 | 0 | |
| muYTransition integer |
Transition rate on Y | muYTransition 0.000001 | 1e-08 | |
| muYTransversion integer |
Transversion rate on Y | muYTransversion 0.0000001 | 0 | |
| muAutosomeTransition integer |
Transition rate on autosomes | muAutosomeTransition 0.000001 | 1e-08 | |
| muAutosomeTransversion integer |
Transversion rate on autosomes | muAutosomeTransversion 0.0000001 | 0 | |
| -mdiv | diversityToOutput 1 |
Output diversity for mitochondrial DNA | ||
| -xdiv | diversityToOutput 2 |
Output diversity for X chromosome | ||
| -ydiv | diversityToOutput 3 |
Output diversity for Y chromosome | ||
| -adiv | diversityToOutput 4 |
Output diversity for autosomes | ||
| diversityToOutput XXX |
XXX can be a list of the above numbers, for outputting diversity of the four types XXX = 0 | diversityToOutput 1 2 3 4 | 0 | |
| -nbLociA integer | nbLociA integer |
Number of unlinked autosomal loci | nbLociA 1 | 1 |
| -sizeA integer | sizeA integer |
Size, in number of sites, per autosomal locus | sizeA 3200 | 3200 |
| -nbLociX integer | nbLociX integer |
Number of unlinked loci on the X chromosome | nbLociX 1 | 1 |
| -sizeX integer | sizeX integer |
Size, in number of sites, per locus on the X chromosome | sizeX 3200 | 3200 |
| -sizeY integer | sizeY integer |
Size, in number of sites, of the Y chromosome | sizeY 3200 | 3200 |
| -sizeMt integer | sizeMt integer |
Size, in number of sites, of the mitochondrial sequence | sizeMt 3200 | 3200 |
| -burnin double | burninTheta double |
Set the diversity threshold parameter for the burn-in phase | burninTheta 0.5 | 0 |
| -burninType int | burninType int |
Set the type of DNA to be tested for the burn-in phase | burninType 1 | 1 |
| -demog double(x3) | demog double (x3) |
Set the demographic function popsize(t+1) = a + b × popsize(t) + c × popsize(t)2 | demog 0 1 0 | 0 1 0 |
| -fasta | fastaOuput integer (0 or 1) |
Save each simulation in a fasta format file at the end of the run | fastaOutput 1 | 0 |
| -arl | arlequinOutput integer (0 or 1) |
Save each simulation in a arlequin format file at the end of the run | arlequinOuput 1 | 0 |
| -save string | save integer (0 or 1) (+string) |
save all the simulations in the SMARTPOP format (.sim) in the directory given by the argument | save 1 mydirectory | 0 |
| -load string | load integer (0 or 1) (+string) |
load all the simulations in a SMARTPOP format (.sim) from the directory given in argument | load 1 mydirectory | 0 |
| -header | headerOutput integer (0 or 1) |
Output the header at the beginning of diversity files | headerOutput 1 | 0 |
B References
[1] P Donnelly and S Tavaré. Coalescents and genealogical structure under neutrality. Annual Review of Genetics, 29(1):401–421, 1995.
[2] Laurent Excoffier and Heidi E L Lischer. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3):564–567, May 2010.
[3] Richard R Hudson. Gene genealogies and the coalescent process. Oxford Surveys in Evolutionary Biology, 7(1):44, 1990.
[4] Masatoshi Nei. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 89(3):583–590, 1978.
[5] K Thornton. LibSequence: A C++ class library for evolutionary genetic analysis. Bioinformatics, 19(17):2325–2327, 2003.
[6] John Wakeley. Coalescent Theory: An Introduction. Roberts & Company Publishers, first edition, 2009.