
Examples¶
We present here two examples of analyses using SMARTPOP. The bash scripts and R code can be downloaded here.
More examples are available in the original paper: SMARTPOP: inferring the impact of social dynamics on genetic diversity through high speed simulations by Guillot and Cox, BMC Bioinformatics 2014 15:175.
Equilibrium state of diversity depending on the population size
Comparing the dynamics of diversity between monogamous and polygamous populations
Equilibrium state of diversity depending on the population size¶
In this example, we run 500 simulations (--nsimu 500) with a population of 100 individuals (-p 100) having a polygamous mating system (--polygamy 3, i.e. up to 3 wives per men). We measure diversity estimators for the population after a long run when the population has reached equilibrium (-t 10000). We check the diversity on a sample of 50 individuals (--sample 50). The output will be named example1 (-o example1), and includes the header for the analysis (--header)
./smartpop -p 100 --polygamy 3 -t 10000 --sample 50 --header -o example1
To produce datasets for higher population sizes, we just repeat the same command with the desired population size. We will append the results to the same file. The header should not be added each time, so we must get remove the --header flag.
./smartpop -p 500 --polygamy 3 -t 10000 --sample 50 -o example1
./smartpop -p 1000 --polygamy 3 -t 10000 --sample 50 -o example1
Using an R script, we can look at the diversity at equilibrium depending on the population size.
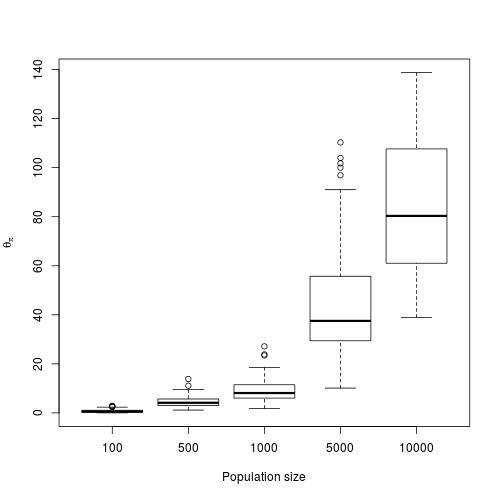
Simulated θπ depending on the population size.
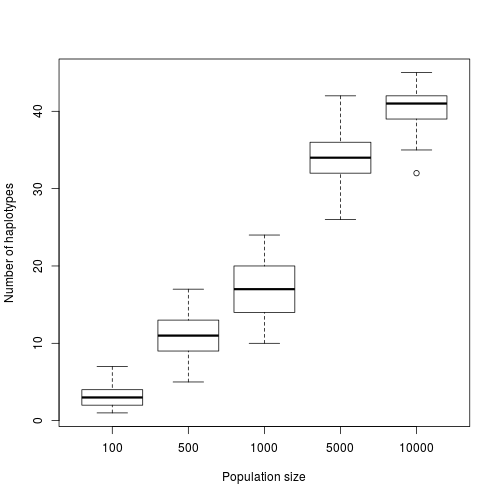
Simulated number of haplotypes depending on the population size.
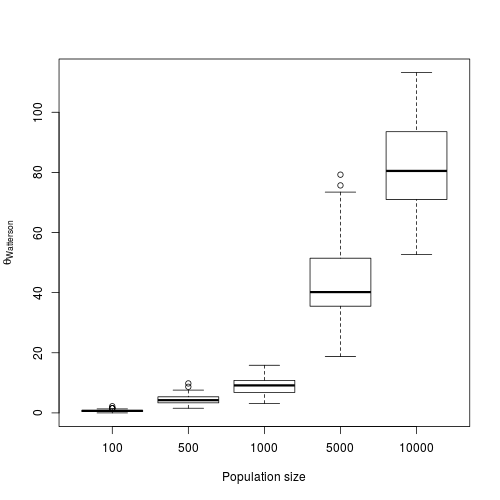
Simulated θWatterson depending on the population size.
Comparing the dynamics of diversity patterns between monogamy and polygamy¶
In this example, we run 1000 simulations (--nsimu 1000) with a population of 200 individuals (-p 200) having a monogamous system (--polygamy 1) with no sibling alliance (--inbreeding 1). This simulation uses a burn-in phase to first increase the population diversity until θπ reaches 25 (--burnin 25). We then follow different estimators in the population through time. The diversity is checked on a sample of 20 individuals (--sample 20) every 5 generations (-t 5), 50 successive times (--nstep 50). The output files will be named example2_mono (-o example2_mono) and include the header for the analysis (--header).
The complete command line to produce the monogamous simulations is:
./smartpop -p 100 --nsimu 1000 --polygamy 1 --sample 20 --inbreeding 1 -t 5 --nstep 50 -o example2_mono --header
The complete command line to produce the polygamous simulations is:
./smartpop -p 100 --nsimu 1000 -polygamy 5 --sample 20 --inbreeding 1 -t 5 --nstep 50 -o example2_poly --header
This will create the following file:
- example2_monoall.txt: Estimators on simulated DNA through time under monogamy.
- example2_polyall.txt: Estimators on simulated DNA through time under polygamy.
Those outputs can be analyzed using R (cf. example2.R).
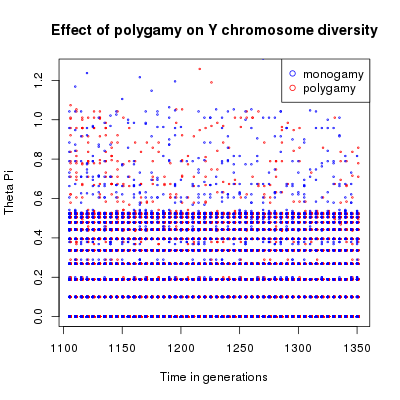
θπ through time. Red points represent monogamous populations, blue points represent polygamous populations.
It is more readable to measure the mean of simulations having the same mating system at the same time point.
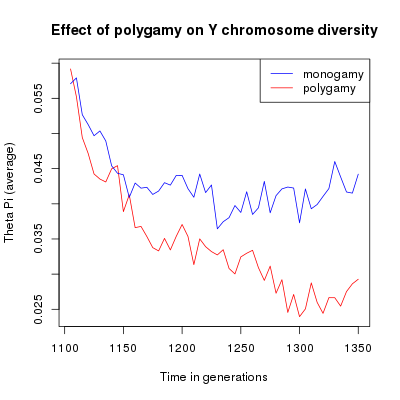
Average θπ through time. The red line represents monogamous populations, the blue line represents polygamous populations.
It is interesting to follow the number of haplotypes on the same simulation set.
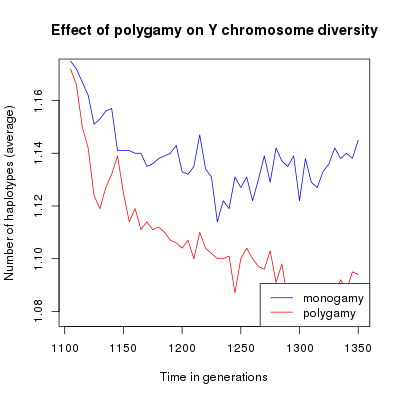
Average number of haplotypes through time. The red line represents monogamous populations, the blue line represents polygamous populations.
With this example, looking at average values you can see that the mating system introduce a difference in the population genetic diversity using both estimators. This difference is however quite small. The scatter plot show a large variance between simulations which makes the difference between monogamy and polygamy non significant.